

解码生命调控密码|EMSA & ChIP 蛋白-核酸互作检测黄金方案
在基因表达的精密蓝图中,蛋白与核酸的互作是解码生命调控的核心密码。从疾病机制的分子溯源到药物靶点的精准解析,揭开二者结合模式的核心机制,正是您突破科研瓶颈的关键。
如何精准捕获这对“生命密钥”的互动轨迹?
EMSA与ChIP技术,作为解析蛋白 - 核酸互作的黄金双钥,将通过分子层面的动态“抓拍”与基因组级的全景“测绘”,为您构建从机制假设到功能验证的完整证据链。
﹝EMSA﹞凝胶迁移或电泳迁移率实验
1. EMSA 技术原理
基于蛋白质与带负电荷的核酸结合后,形成的复合物在非变性聚丙烯酰胺凝胶(PAGE)或琼脂糖凝胶中电泳迁移速度变慢的原理。游离的核酸探针跑得快(在凝胶下方),而蛋白-核酸复合物因分子量增大和电荷被中和而迁移慢(在凝胶上方形成“条带偏移”)。
EMSA 实验根据实验方案设计的不同,分为验证型 EMSA、竞争型 EMSA、超迁移 EMSA。
|
EMSA 类型 |
验证型 EMSA | 竞争型 EMSA | 超迁移 EMSA |
|---|---|---|---|
| 核心目的 | 检测蛋白质是否能与目标核酸探针结合 | 评估结合的序列特异性 | 鉴定复合物中的特定蛋白质组分 |
| 关键设计要素 | 标记探针 + 递增浓度蛋白提取物 + 阴性对照 | 标记探针 + 固定量蛋白 + 递增浓度未标记竞争剂 | 标记探针 + 蛋白形成复合物 + 加入特定抗体 |
| 预期结果 | 出现迁移变慢的条带(shift),强度随蛋白浓度增加而增强 | 同源竞争剂:复合物条带减弱/消失 突变竞争剂:复合物条带基本不变 |
出现迁移更慢的新条带(supershift),原复合物条带可能减弱/消失 |
| 验证点 | 结合能力的存在 | 结合的序列特异性 | 特定蛋白存在于复合物中 |
2. EMSA 应用场景
转录调控解析:精准鉴定转录因子与 DNA/RNA 顺式作用元件的结合位点,绘制基因表达调控蓝图。
药物研发加速:筛选抑制蛋白 - 核酸结合的小分子化合物,为靶向药物开发提供关键数据支撑。
疾病机制探索:分析基因突变对转录因子结合的影响,如癌症中调控通路的异常激活机制研究。
特点: 相对简单、快速、直观(直接看到条带偏移),是验证蛋白质-核酸相互作用的经典“金标准”方法之一。适用于体外研究,灵敏度取决于探针标记方式。
3. EMSA 常见问题及解决方案
|
问题 |
可能原因 |
解决方案 |
|
探针标记效率低或无标记 |
标记酶活性降低:T4 多聚核苷酸激酶等酶保存不当(如反复冻融)。 ATP / 标记物失效:放射性 α-32P-ATP 或荧光标记 dNTP 超过保质期。 反应体系错误:缓冲液成分遗漏(如 Mg²⁺)或探针浓度过高 / 过低。 |
酶活性验证:使用新鲜分装的酶,避免反复冻融,实验前用已知探针做阳性对照。 试剂更换:检查 ATP / 标记物的有效期,重新配制或购买新试剂。 优化体系:严格按照试剂盒说明书调整探针浓度(通常 10-50 ng),确保缓冲液成分完整。 延长反应时间:标记反应可在 37℃延长至 60 分钟,或增加酶的用量。 |
|
电泳后无条带或条带模糊 |
蛋白质与探针结合效率低:蛋白质浓度不足或变性失活(如未使用蛋白酶抑制剂);探针未纯化,含杂质抑制结合。 电泳条件不当:凝胶浓度不合适(如 DNA 探针常用 4%-6% 聚丙烯酰胺凝胶,RNA 需更高浓度);电泳缓冲液失效(TBE 或 TAE 缓冲液重复使用次数过多)。 非特异性结合干扰:未添加足够的非特异性竞争 DNA(如 Poly (dI・dC))。 |
优化蛋白质制备:使用新鲜提取的蛋白质,添加蛋白酶抑制剂(如 PMSF)防止降解;通过预实验确定最佳蛋白浓度(建议梯度测试 50-500 ng)。 探针纯化:标记后通过乙醇沉淀或柱纯化去除未结合的标记物和杂质。 调整电泳参数:根据探针长度选择凝胶浓度(短探针用高浓度,长探针用低浓度);更换新鲜配制的 1× TBE 缓冲液,电泳温度控制在 4℃(避免核酸变性)。 增加竞争剂:非特异性竞争 DNA 的用量通常为探针的 50-100 倍,特异性竞争 DNA 为 10-100 倍。 |
|
非特异性条带过多 |
竞争剂不足:未添加或用量不够,导致蛋白质与探针非特异性结合。 探针浓度过高:过量探针与蛋白质非特异性结合。 反应缓冲液成分问题:缺乏 Mg²⁺、Zn²⁺等辅助因子,或盐浓度过高抑制结合。 |
优化竞争条件:增加非特异性竞争 DNA(如 Poly (dI・dC))的用量,或加入载体蛋白(如 BSA)减少非特异性吸附。 降低探针浓度:将探针浓度降至 1-5 nM,避免过量结合。 调整缓冲液:补充 MgCl₂(1-5 mM)或 ZnCl₂(0.1-1 mM)促进蛋白质 - DNA 结合;降低 NaCl 浓度(通常 10-100 mM),过高盐浓度会破坏结合力。 特异性验证:加入抗体进行超迁移实验(Super Shift),确认特异性条带(抗体 - 蛋白 - DNA 复合物迁移率更低)。 |
|
条带迁移率异常 |
蛋白质聚合或降解:蛋白质形成多聚体导致迁移率降低,或降解为小片段导致迁移率升高。 探针二级结构影响:探针自身形成发夹结构,影响与蛋白质的结合及迁移率。 凝胶交联度异常:丙烯酰胺与 bis 丙烯酰胺比例错误(通常 29:1),导致凝胶孔径不均。 |
蛋白质状态优化:用 Native-PAGE 检测蛋白质纯度,避免聚合或降解(可添加 10% 甘油稳定蛋白质);若蛋白质易聚合,可在反应体系中加入 0.1% NP-40 等去垢剂破坏聚合体。 探针预处理:标记前对探针进行变性 - 复性处理(95℃加热 5 分钟,冰浴退火),消除二级结构。 重新配制凝胶:严格按照丙烯酰胺与 bis 丙烯酰胺比例(29:1)配制,确保凝胶均匀凝固。 |
|
曝光/成像信号弱或背景高 |
探针标记效率低:同 “探针标记效率低” 问题。 洗膜 / 洗涤不充分:未结合的探针残留导致背景高(尤其荧光检测时)。 检测条件不当:放射性曝光时间过短,或荧光成像仪参数设置错误(如曝光时间、增益过高)。 |
提高标记效率:参考 “探针标记效率低” 的解决方案,确保标记后探针比活性足够。 优化洗涤步骤: 放射性实验:电泳后用 0.5× TBE 缓冲液洗胶 10-15 分钟,去除游离探针。 荧光实验:用含 0.1% Tween-20 的 PBST 缓冲液洗涤 3 次,每次 5 分钟。 调整检测参数: 放射性曝光:延长曝光时间(如从 12 小时增至 24 小时),或使用增感屏增强信号。 荧光检测:降低激发光强度或曝光时间,避免背景过曝。 |
|
重复实验结果不一致 |
试剂批次差异:蛋白质提取物、探针或缓冲液批次不同,导致结合效率不稳定。 操作误差:反应体系配制时体积误差、凝胶厚度不均一或电泳温度波动。 蛋白质活性变化:冻融次数过多导致蛋白质活性下降。 |
标准化试剂:使用同一批次的蛋白质提取物,分装保存避免反复冻融(-80℃保存,每次用新鲜分装);探针标记后定量浓度,确保每次实验用量一致。 规范操作:使用微量移液器精确配制反应体系,避免气泡;制胶时确保玻璃板水平,凝固时间一致(通常 30-60 分钟);电泳过程中保持温度稳定(可在冷室中进行)。 设置内对照:每次实验加入已知结合的阳性对照(如 SP1 与 DNA 探针),验证体系稳定性。 |
其他注意事项(预防常见问题):
实验耗材处理:使用 DEPC 水处理玻璃器皿(RNA-EMSA),或硅化处理避免核酸吸附。
蛋白质 - 核酸结合条件:根据蛋白质特性调整 pH(通常 7.0-8.0)和温度(室温或 37℃)。
预实验优化:通过梯度实验确定最佳蛋白浓度、探针浓度和竞争剂用量。
安全防护:放射性标记实验需穿戴防护装备,废弃试剂按放射性废物处理。
﹝ChIP﹞染色质免疫沉淀
1. ChIP 技术原理
在活细胞状态下,利用甲醛等交联剂将细胞内蛋白质与DNA(或蛋白质与蛋白质-DNA复合物)“固定”在一起。裂解细胞,将染色质随机打断成小片段。用针对目标蛋白质的特异性抗体进行免疫沉淀,将与目标蛋白交联在一起的DNA片段富集下来。解除交联后,纯化富集的DNA片段,进行下游分析。
ChIP 技术根据染色质片段化方式和是否使用甲醛交联可分为交联型ChIP(X-ChIP)、天然型ChIP(N-ChIP)两大核心路线:
| 特征 | X-ChIP(交联型) | N-ChIP(天然型) |
|---|---|---|
| 交联剂 | 甲醛(固定瞬时互作) | 无 |
| 片段化方法 | 超声破碎(机械力) | 微球菌核酸酶MNase(酶切) |
| 适用目标 | 转录因子、动态结合蛋白 | 组蛋白修饰、稳定结合蛋白 |
| 分辨率 | 200~1000 bp(依赖超声条件) | 核小体水平(~147 bp) |
| 特异性背景 | 较高(交联可能引入非特异性结合) | 较低(保留天然互作) |
| 关键优化点 | 交联时间、超声强度 | MNase浓度、消化时间 |
选择路线的决策逻辑:
-
研究转录因子? → 必选 X-ChIP(因子结合短暂,需交联固定)
-
研究组蛋白修饰? → 优先 N-ChIP(高分辨率、低背景,但需验证目标修饰是否稳定)
-
研究染色质结合蛋白的稳定性未知? → 可尝试 X-ChIP(兼容性更广,但需严谨对照)
2. ChIP 应用场景
转录调控网络构建:绘制转录因子在全基因组的结合图谱,揭示基因表达的全局调控机制。
表观遗传学研究:分析组蛋白修饰与基因激活 / 沉默的关系,探索染色质状态对基因功能的影响。
疾病靶点发现:筛选癌症等疾病中异常激活的转录因子结合位点,为靶向治疗提供新方向。
特点: 反映细胞内、生理状态下的真实结合情况,是研究基因组层面蛋白质-DNA相互作用的金标准。技术难度相对EMSA更高,步骤繁琐,成本较高(尤其ChIP-seq),对抗体特异性要求极高(是实验成败的关键)。
3. ChIP 常见问题及解决方案
|
问题 |
可能原因 |
解决方案 |
|
背景高 / 非特异性结合严重 |
抗体特异性不足:抗体识别非目标蛋白,或与 DNA 非特异性结合。 超声破碎不充分:染色质片段过大(>1000 bp),导致非特异性沉淀。 封闭不彻底:磁珠 / 琼脂糖珠未用 BSA 或鲑鱼精 DNA 充分封闭。 洗涤不充分:未有效去除非特异性结合的染色质片段。 |
验证抗体特异性:使用 Western blot 预验证抗体对目标蛋白的识别能力;选择 ChIP 级抗体(注明适用于 ChIP 实验),避免使用 IHC 或 WB 专用抗体。 优化超声条件:调整超声功率和时间(如 200 W,每次 30 秒,共 10 次),目标片段长度控制在 200-500 bp;超声时保持样本在冰浴中,防止蛋白质变性。 增强封闭效果:磁珠封闭液中添加 5% BSA 和 100 μg/mL 鲑鱼精 DNA,室温孵育 30 分钟;避免使用脱脂奶粉(含 DNA 酶),改用 BSA 作为封闭剂。 严格洗涤步骤:增加洗涤次数(如 5 次),使用含高盐(150-500 mM NaCl)和 Triton X-100 的洗涤缓冲液;最后一次洗涤用低盐缓冲液(10 mM Tris-HCl, pH 8.0)去除残留盐离子。 |
|
目的基因富集效率低 |
交联不充分:甲醛浓度不足或交联时间过短,蛋白质 - DNA 复合物解离。 抗体量不足:抗体浓度过低,无法有效捕获目标蛋白。 染色质片段丢失:超声后碎片沉淀或操作中损失。 PCR 效率问题:引物设计不合理或 qPCR 体系污染。 |
优化交联条件:常用 1% 甲醛室温交联 10-15 分钟,加甘氨酸(终浓度 125 mM)终止反应;贴壁细胞交联时需确保甲醛均匀接触,悬浮细胞需充分重悬。 调整抗体用量:预实验测试抗体浓度(如 5-10 μg/107 cells),避免过量(可能导致非特异性结合);加入抗体后 4℃旋转孵育过夜,增强结合效率。 减少样本损失:超声后离心(12000 g, 10 分钟)取上清,避免吸入沉淀的细胞碎片;磁珠结合时轻柔颠倒混合,避免剧烈涡旋导致磁珠破碎。 优化 PCR 体系:设计跨核小体的引物(扩增片段 80-150 bp),用 Input DNA 验证引物效率;使用 ChIP 级 qPCR 试剂盒,避免外源 DNA 污染(实验器材高压灭菌,戴手套操作)。 |
|
DNA 片段降解严重 |
超声过度:功率过高或时间过长,导致 DNA 碎片化。 核酸酶污染:实验器材或试剂含 DNA 酶 / RNA 酶。 交联过度:甲醛浓度过高或交联时间过长,DNA - 蛋白质交联难以逆转。 |
优化超声参数:采用脉冲式超声(如开 5 秒,停 10 秒),降低功率(150-200 W),分多次短时间处理;超声前取少量样本检测片段大小,调整至 200-500 bp 区间。 防核酸酶污染:所有器材经 DEPC 水处理或高压灭菌,试剂中添加核酸酶抑制剂(如 RNase Inhibitor);操作时戴手套,避免手汗中的核酸酶污染样本。 控制交联程度:交联时间不超过 20 分钟,甲醛浓度不超过 2%,必要时用甲醇固定细胞(适用于易交联蛋白);逆转交联时 65℃孵育 4-6 小时,或添加 NaCl(终浓度 200 mM)促进交联逆转。 |
|
样本间重复性差 |
细胞数量不一致:不同样本细胞数差异超过 20%,导致蛋白量不均一。 操作误差:抗体孵育、洗涤或 DNA 提取步骤时间控制不一致。 试剂批次差异:抗体、磁珠或缓冲液批次不同,性能不稳定。 |
标准化细胞处理:用台盼蓝染色计数活细胞,每样本使用相同数量细胞(如 107 cells);处理贴壁细胞时用胰酶消化后重悬,避免机械刮擦导致细胞损伤。 规范实验操作:制定详细操作时间表,确保各步骤孵育时间一致(如抗体孵育 16 小时,洗涤每次 5 分钟);磁珠分离时使用磁力架固定时间(如 5 分钟),避免手动倾倒导致损失。 统一试剂批次:抗体和磁珠购买同一批次,分装保存(4℃或 - 20℃),避免反复冻融;缓冲液提前配制并分装,使用前平衡至室温。 |
|
qPCR 结果异常(如 Ct 值过高或无扩增) |
DNA 模板量不足:ChIP 富集效率低,或 DNA 提取过程中损失。 引物二聚体或污染:引物设计导致非特异性扩增,或体系被外源 DNA 污染。 逆转交联不彻底:DNA - 蛋白质交联未完全打开,抑制 PCR。 |
增加模板量:浓缩 ChIP DNA(如乙醇沉淀),或增加 qPCR 反应中模板体积(不超过体系的 1/10);确保 Input DNA 稀释度一致(如 1:100),作为阳性对照。 优化引物设计:使用 Primer3 等工具设计引物,避免 GC 含量过高(<60%)或发卡结构,Tm 值控制在 58-62℃;用蒸馏水做阴性对照,检测是否存在试剂污染。 彻底逆转交联:逆转交联时加入 NaCl(终浓度 500 mM)和 RNase A(100 μg/mL),65℃孵育过夜;必要时添加 Proteinase K(200 μg/mL)消化蛋白质,确保 DNA 完全释放。 |
|
ChIP-seq 数据质量低(如峰不明显) |
抗体富集效率低:目标蛋白在基因组上结合位点少,或抗体识别表位被交联掩盖。 测序深度不足:文库测序数据量不够(如 < 1000 万 reads),无法检测低丰度结合位点。 背景噪声高:IgG 对照信号强,掩盖真实结合信号。 |
验证结合位点:先用 ChIP-qPCR 验证已知结合位点(如启动子区域),确认实验体系有效性;若目标蛋白为转录因子,可参考 ENCODE 数据库预测结合位点,缩小检测范围。 增加测序数据量:提高测序深度至 1500 万 - 2000 万 reads,或使用高灵敏度测序试剂盒(如 Nugen Ovation);对 ChIP DNA 进行两轮 PCR 扩增,提高文库浓度(但需控制循环数≤18 次,避免偏差)。 优化数据处理:使用 MACS2 等软件分析数据时,提高峰识别阈值(如 q-value < 0.01),过滤低质量峰;对比 Input 样本扣除背景,或使用多个 IgG 对照平均化背景信号。 |
|
特殊样本处理问题(如原代细胞、组织) |
组织解离不充分:细胞未完全分散,导致交联和超声不均匀。 细胞异质性:组织中目标细胞比例低,蛋白丰度不足。 样本量不足:原代细胞培养数量有限,难以获得足够染色质。 |
优化组织处理:新鲜组织用剪刀剪碎后,加入胶原酶 / 胰酶混合液 37℃消化 30-60 分钟,过筛获得单细胞悬液;解离后用 PBS 洗去消化酶,避免酶残留降解蛋白质。 富集目标细胞:通过流式细胞术(FACS)或磁珠分选(MACS)纯化目标细胞群(如 CD4+ T 细胞);若目标细胞比例低,可增加组织用量(如 500 mg 组织对应 107 cells)。 调整实验体系:原代细胞 ChIP 可减少细胞用量至 5×106 cells,增加抗体浓度(如 10 μg/5×106 cells);组织样本超声时降低功率(100-150 W),延长脉冲间隔,避免细胞碎片堵塞超声探头。 |
其他注意事项(预防常见问题):
阳性 / 阴性对照设置:
阳性对照:使用已知结合的抗体(如 RNA Pol II)和引物(如 GAPDH 启动子)。
阴性对照:IgG 同型抗体、无抗体对照(Mock IP),以及非靶基因引物。
样本保存策略:
细胞交联后可冻存于 - 80℃(用 10% DMSO 保护),避免反复冻融影响染色质结构。
超声后的染色质可分装保存,每次使用新鲜分装,减少核酸酶降解。
试剂兼容性:
避免使用含 SDS 的缓冲液(>0.1%),可能破坏抗体 - 抗原结合。
磁珠洗涤时用低转速(如 500 g)离心,防止磁珠聚集沉淀。
EMSA VS ChIP
| 维度 | EMSA | ChIP |
|---|---|---|
| 结合特异性 | 需通过竞争实验验证 | 依赖抗体特异性,需阴性 / 阳性对照 |
| 分辨率 | 可定位至核酸序列中的具体结合位点 | ChIP - seq 可定位至全基因组单碱基水平 |
| 样本要求 | 蛋白提取物或纯化蛋白 + 核酸探针 | 细胞或组织(需保持交联效率) |
| 应用侧重 | 验证已知蛋白 - 核酸的结合及亲和力 | 筛选全基因组范围内的结合位点 |
选择策略:
已知靶点验证:若需验证特定蛋白与核酸序列的结合及亲和力,EMSA 凭借体外操作简便、成本可控的优势,是高效首选。
未知靶点筛选:若致力于探索体内生理状态下的全基因组结合位点,或解析表观遗传调控机制,CHIP 技术(尤其是 CHIP - seq)能提供全景式的调控信息。
代轩生物提供蛋白与核酸检测技术服务!
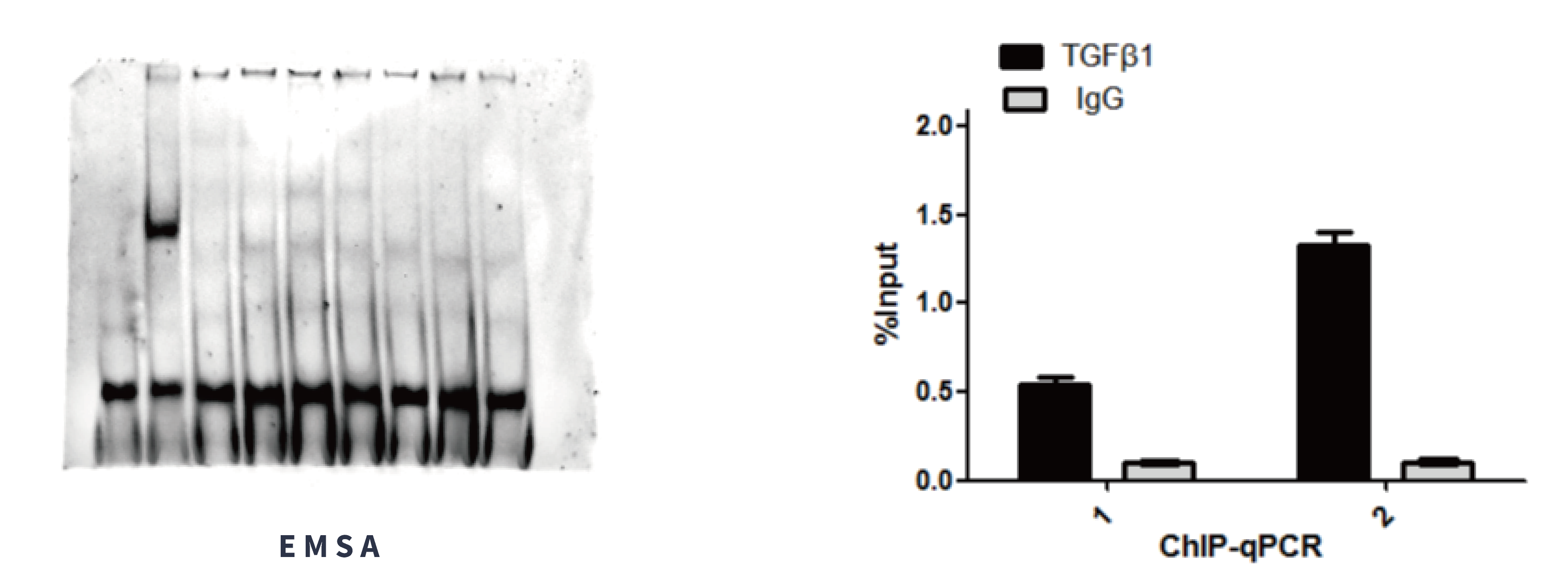
服务内容:
实验设计咨询:根据研究目的推荐最合适的技术方案。
样品处理:核酸探针设计合成与标记(放射性/荧光/生物素)、蛋白质表达纯化(重组蛋白)或细胞/组织裂解物制备。
实验操作:严格按照标准流程进行结合反应、电泳(EMSA)、免疫沉淀(ChIP, Pull-down)、洗涤、检测等。
检测与成像:使用凝胶成像系统(EMSA)、qPCR仪(ChIP-qPCR)、测序仪(ChIP-seq)、质谱仪(Pull-down-MS)、SPR/BLI仪器等。
数据分析与报告:提供原始数据、图像、定量分析结果(如结合百分比、富集倍数、亲和力常数)和实验报告。

